There has been a hard stop on posting over the last several months because we were defending our dissertation work, trying to get manuscripts published, starting our postdocs in a new city, and planning a wedding… After a little bit of stress, and even less sleep, my wife and I were able to successfully complete all of these important life milestones together.

Now that we a both onto the next step, I want to take a second to discuss some work that came from my thesis that I’m really proud of. In a nutshell, through a combination of -omics technologies and genome-scale computational modeling, we were able to identify differences in the nutrient niche of C. difficile across separate antibiotic-pretreatment mouse models of infection that correlated with changes in disease severity. What this means is that which antibiotic you are treated with that sensitizes you to C. difficile colonization or what your personel gut community looks like may effect how severely the pathogen impacts you. The article can be found open-access here. This work was also featured as the Editor’s Pick for July 2017 (vol. 2 (4)) and my ternary plot was selected for that issue’s cover! Here was the winning figure:
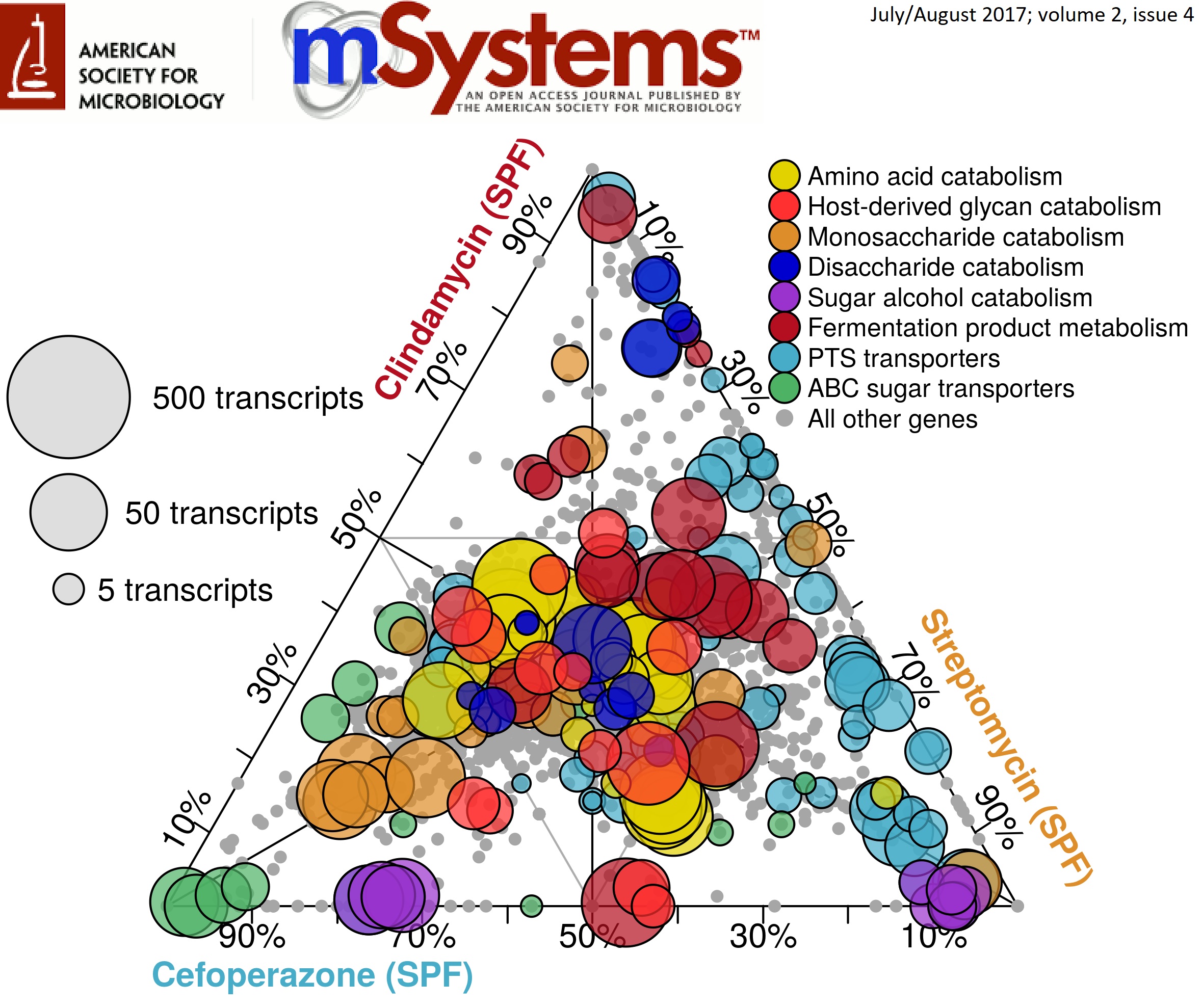
What this figure is showing is the transcript abundance of selected genes in several metabolic pathways across 3 antibiotic pretreatment models. Position of each point reflects the mRNA quantified from each condition associated with each corner. The closer a point is to one edge, the more overrepresented expression of that gene is in that pretreatment group. Size of the points demonstrates the normalized number of transcripts found for that gene in the condition where it’s expression was highest. Overall, this implies that certain forms of metabolism are more active under certain environmental conditions which inform downstream virulence expression. We later confirmed this trend using a simplified metabolic model and untargeted mass spectrometry of metabolites found in the gut during infection.
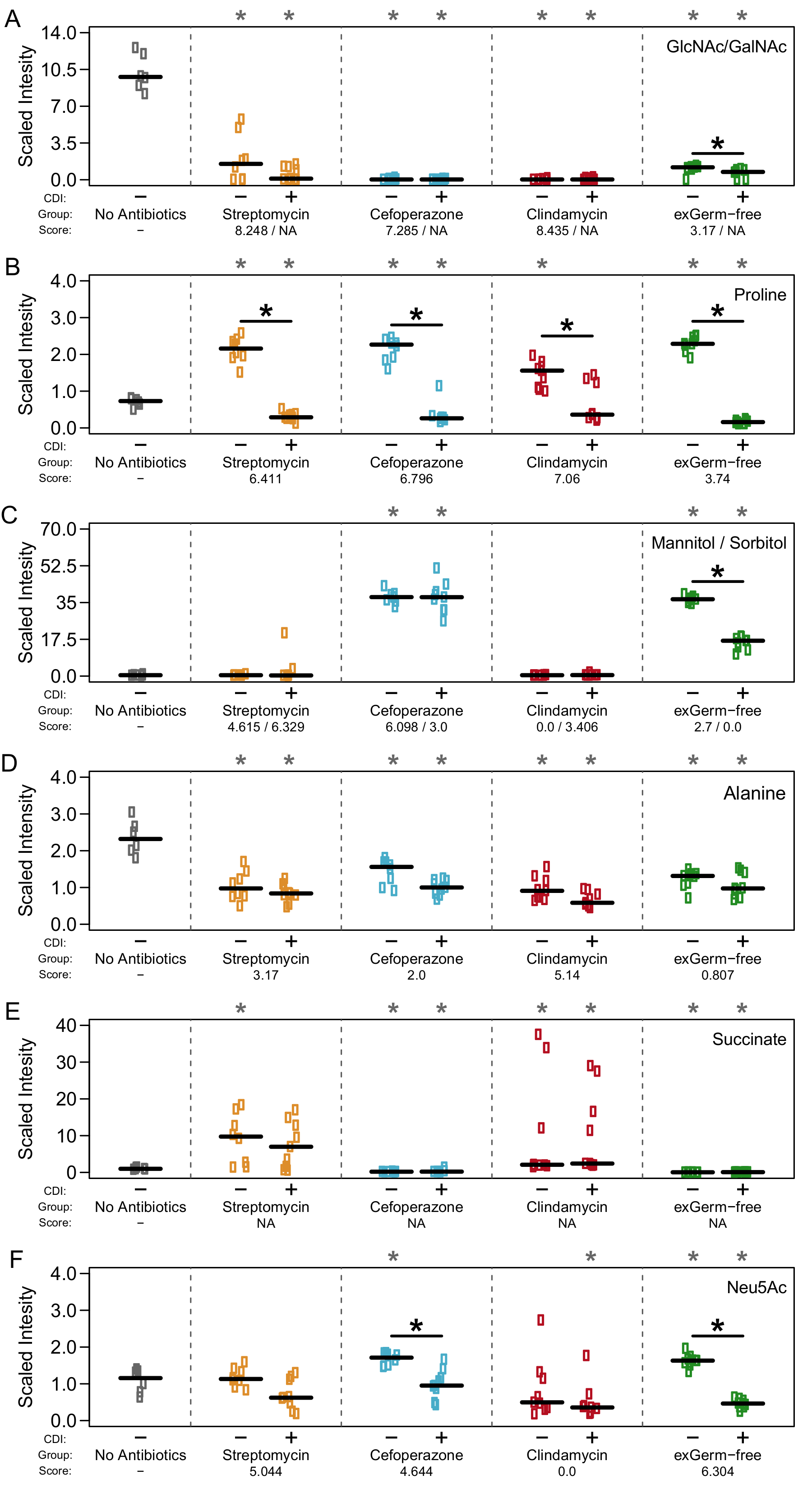
Metabolomic results supported that there was indeed a heirarchy to nutirent preference of C. difficile during infection and this may be determined by which competitors are left over after antibiotics. Check out the rest here!.

