We were recently asked to contribute a review to Current Opinion in Microbiology on computational approaches that have been used to understand different elements of Clostridioides difficile metabolism and virulence. A lot of really great work has been done recently around this topic by our group and others, and this gave a chance to showcase a large selection. I’m really happy with how it turned out, and you can read it here!
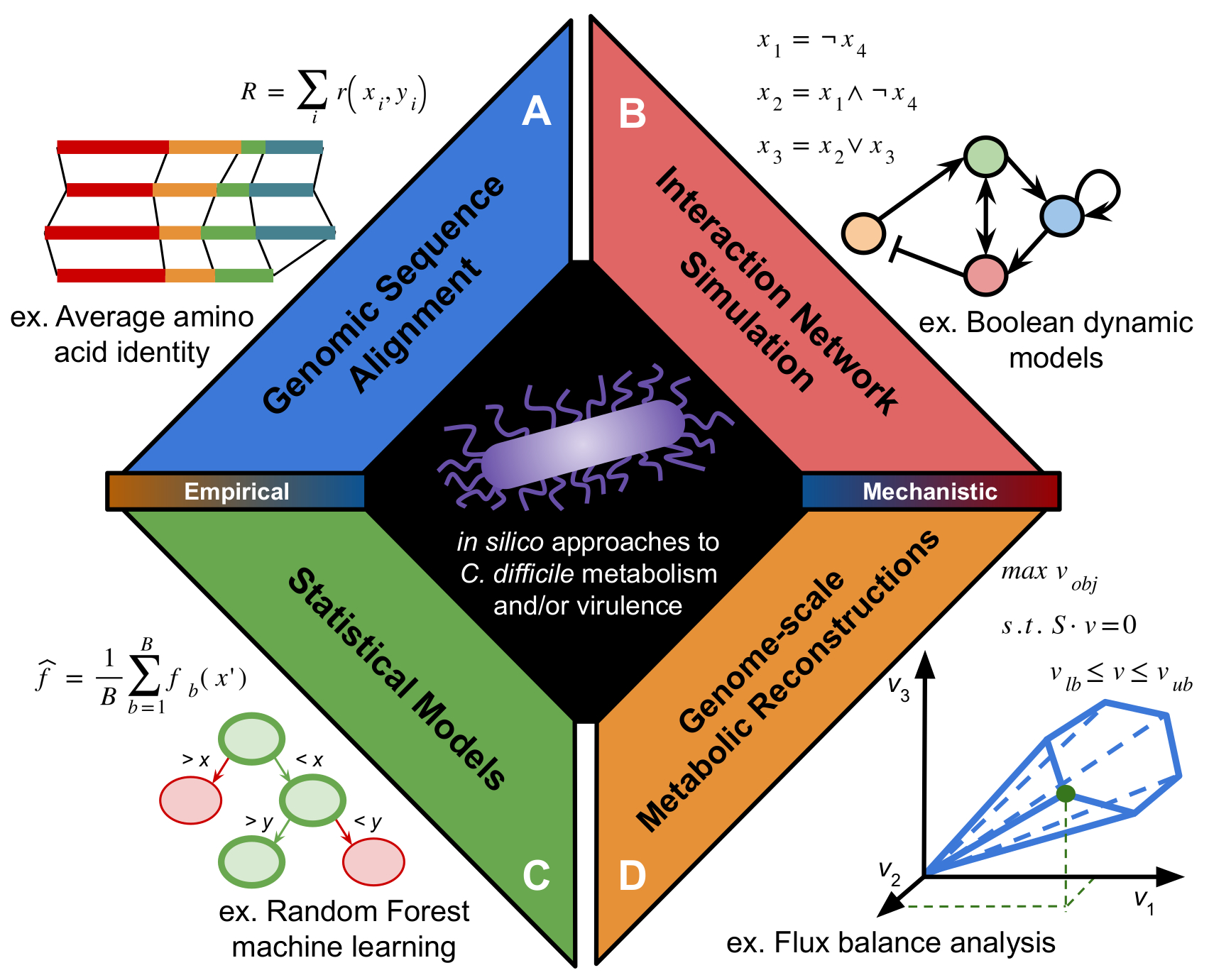
The progress of infection by C. difficile is strongly influenced by metabolic cues it encounters as it colonizes the gastrointestinal tract. Both colonization and regulation of virulence have a multi-factorial interaction between host, microbiome, and gene expression cascades. While these connections with metabolism have been understood for some time, many mechanisms of control have remained difficult to directly assay due to high metabolic variability among C. difficile isolates and difficult genetic systems. Computational systems offer a means to interrogate structure of complex or noisy datasets and generate useful, tractable hypotheses to be tested in the laboratory. Recently, in silico techniques have provided powerful insights into metabolic elements of C. difficile infection ranging from virulence regulation to interactions with the gut microbiota. In our review, we introduce and provide context to the methods of computational modeling that have been applied to C. difficile metabolism and virulence thus far. The techniques discussed there have laid the foundation for future multi-scale efforts aimed at understanding the complex interplay of metabolic activity between pathogen, host, and surrounding microbial community in the regulation of C. difficile pathogenesis. Below is a figure from it outlining the types of methods that have been applied to this interesting problem:
I’d like to thank the editors Drs. Rita Tamayo and Aimee Shen again for giving us the opportunity to contribute to the special issue!
