My first paper from post-doc is out! Transitioning into from sequence-based analyses into linear-algebra-driven metabolic modeling and flux balance analysis was a challenge, so I’m very proud of this new publication. Our new platform integrates transcriptomic data in genome scale models with repect to maximizing cellular economy in light of the transcriptomic investment made into the enzymes indicated by the data.
Transcriptomic analyses of bacteria have become instrumental to our understanding of their responses to changes in their environment. While traditional analyses have been informative, leveraging these datasets within genome-scale metabolic network reconstructions (GENREs) can provide greatly improved context for shifts in pathway utilization and downstream/upstream ramifications for changes in metabolic regulation. Many previous techniques for GENRE transcript integration have focused on creating maximum consensus with input datasets, but these approaches have been shown to generate less accurate metabolic predictions than a transcript-agnostic method of flux minimization (pFBA), which identifies the most efficient/economic patterns of metabolism given certain growth constraints. Despite this success, growth conditions are not always easily quantifiable and highlights the need for novel platforms that build from these findings. This method, known as RIPTiDe, combines these concepts and utilizes overall minimization of flux weighted by transcriptomic analysis to identify the most energy efficient pathways to achieve growth that include more highly transcribed enzymes, without previous insight into extracellular conditions. This platform could be important for revealing context-specific bacterial phenotypes in line with governing principles of adaptive evolution, that drive disease manifestation or interactions between microbes.
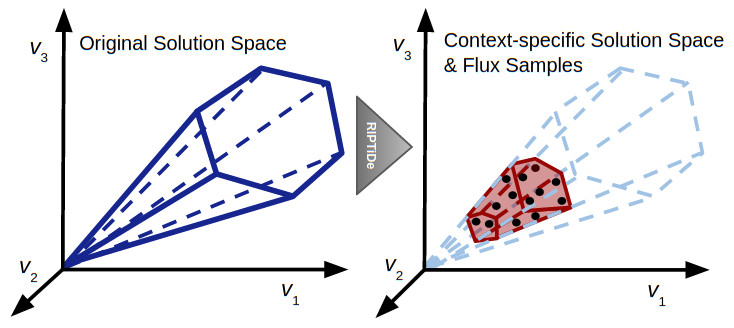
This figure is a representation of the impact of RIPTiDe on the original reconstruction, converting it to a context-specific model of metabolism with a parsimonious metabolic solution space with respect to the given transcriptomic data.
You can read the whole open-access paper here!