Getting rid of duplicate protein sequences becomes important when you are trying to annotate metagenomes. Redundant sequences can compete for reads when mapping to onto genes and don’t add anything extra information when assessing the functional capacity of a group of organisms. Probably the best way to remove identical or highly similar sequences from a FASTA file is through clustering homologous genes. I’ll show some results on data reduction from clustering experiments I ran on a single gene from many organisms and give you my opinion on when, and when not, to be stringent be be when clustering.
The platform I use to do my peptide sequence clustering is CD-HIT. It’s blazing fast and extremely simple to use. Basically, their algorithm clusters sequences based on word count overlap. What this means is that the user specifies a word size, meaning how many consecutive amino acids to consider at a time, and a percent identity as a cutoff on how similar sequences need to be to cluster together. CD-HIT starts by ordering the sequences from longest to shortest, then splitting each sequences into all posible words of the given size. It then counts the number of times each word appears in each sequences, giving each peptide a “signature” of abundances. The benefit of this step is twofold. First, it greatly simplifies each sequence to a small array, reducing the amount of memory needed to process each sequence. It also provides a very simple metric of comparison between peptides. The more their word counts overlap, the more similar they are. This idea was studied more in a study from back in 1998. If sequences fall within the user provided identity, then they are clustered together. Otherwise, a new cluster is formed. This happens processively until all peptides are put either into an existing cluster or make new ones. This procedure is known as greedy incremental clustering.

It’s easy to run CD-HIT once it’s installed. Here’s the command I used:
cd-hit -i metagenome.genes.peptide.fasta -o metagenome.genes.peptide.99 -c 0.99 -n 5 -M 44000 -T 8Here’s what each of the arguments mean:
1
2
3
4
5
6
-i = input FASTA file
-o = output file names
-c = percent identity (99%)
-n = word length
-M = allowed memory usage in Mb (=44 Gb)
-T = number of threads for parallelization
CD-HIT produces 2 files as output: a FASTA file of the first member added to each cluster, and a text file listing which sequences from the original FASTA belong to each cluster.
For a test case, I looked to a paper from synthetic biology. A group used data from systematic deletion experiments to define the minimal list of genes to function as a free-living bacterial genome. What resulted was a list of genes that have some homolog in many, if not all, bacteria. Using one of these genes makes for a great way to test how how many sequences are clustered together as the minimum percent identity is decreased. I chose adenylate kinase (adk), and downloaded the protein sequence for 200 bacterial homologs to a FASTA file from NCBI.
I performed a range of percent identity clusterings from 100% to 70% using the same word size of 5. Below is a summary of the results:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
# Original FASTA
# Total sequences: 200
# Fraction of original: 100%
# Following clustering:
# Percent identity: 100%
# Total sequences: 186
# Fraction of original: 93.0%
# Percent identity: 99%
# Total sequences: 183
# Fraction of original: 91.5%
# Percent identity: 90%
# Total sequences: 178
# Fraction of original: 89.0%
# Percent identity: 80%
# Total sequences: 156
# Fraction of original: 78.0%
# Percent identity: 70%
# Total sequences: 130
# Fraction of original: 65.0%
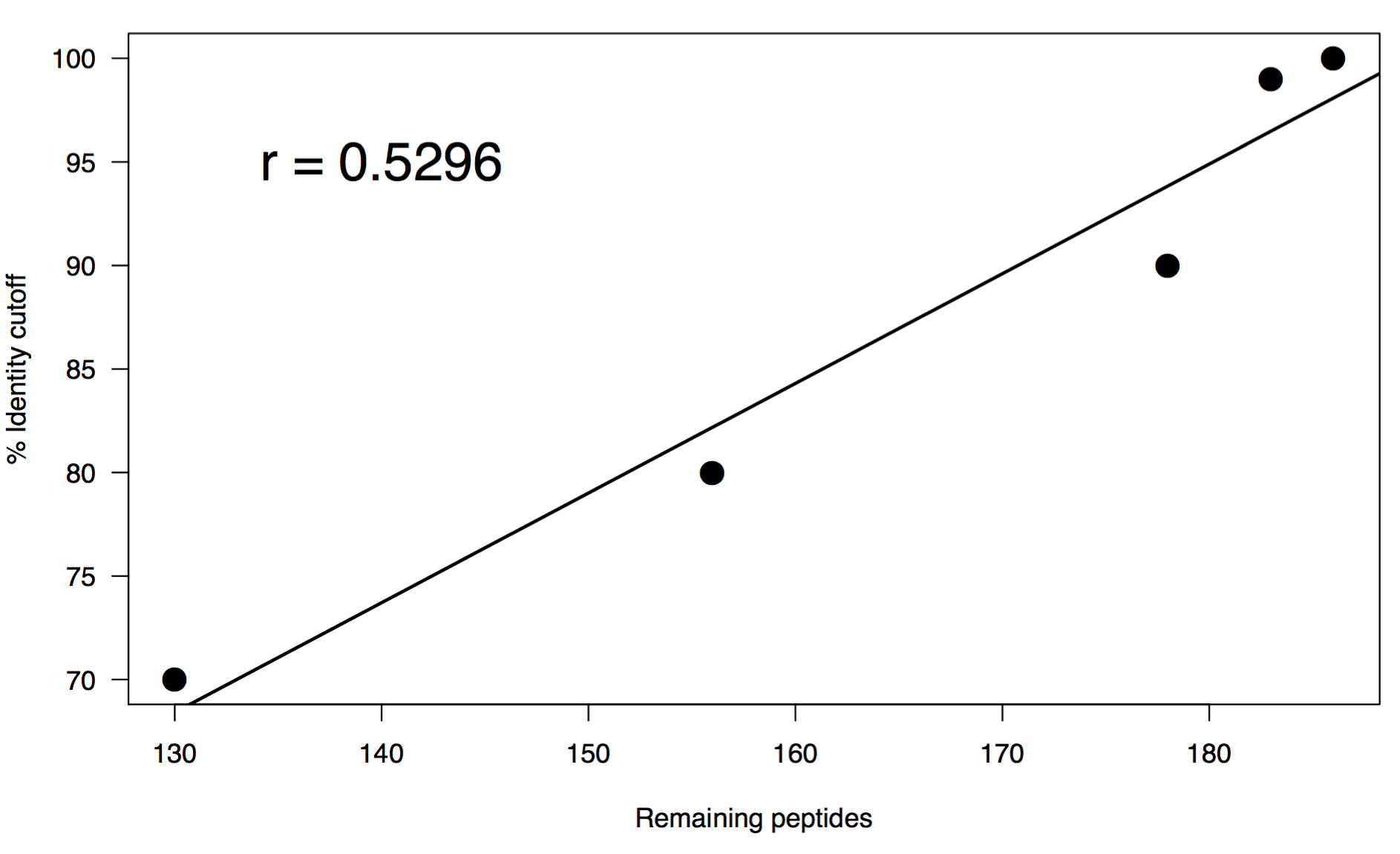
These results are interesting in that there is a pretty swift drop off even clustering at 100%. This may not be all that surprising since they do all come from the exact same gene, but you could also expect a greater level of diversity on adk across species. You may be able to chalk this up to how well the gene is conserved so it’s sequence might only deviate slightly. After this initial droff, there is a linear relationship in the rate at which sequences cluster vs percent identity cutoff. I think that what it comes down to in the end is if you are trying to capture the full repetoire of sequence diversity of homologs in a metagenome, or if you want to look at the relative functional capacity of a collection of genes. Either way, some amount of dereplication is necessary to avoid genes equally competing for reads when you reach the mapping steps of your analyses.