Incidences of infection by K. pneumoniae have grown in frequency to become the leading agents of CRE infection among hospitalized patients in the United States and Europe. Transcriptomic meta-analysis of data collected from both laboratory and clinical isolates indicated significant shifts in expression of key transcription factors related to metabolism. Metabolic network reconstructions have previously proven effective for quickly identifying potential targets in silico, therefore we combined these approaches by integrating the transcriptomic data from each isolate type into a well-curated GENRE of K. pneumoniae to predict emergent metabolic patterns. Leveraging this systems-biology approach we found discordant patterns of active metabolism between clinical and laboratory isolates, with a striking difference in L-valine catabolism. Exogenous valine is known to increase macrophage phagocytosis, and our results may support immunomodulatory activity in K. pneumoniae evolved to avoid host clearance.
Below is a visual representation for the general procedure of generating context-specific models of metabolism from transcriptomic data. All 56 datasets from the transcriptome meta-analysis were used to generate distinct context-specific models of K. pneumoniae metabolism.
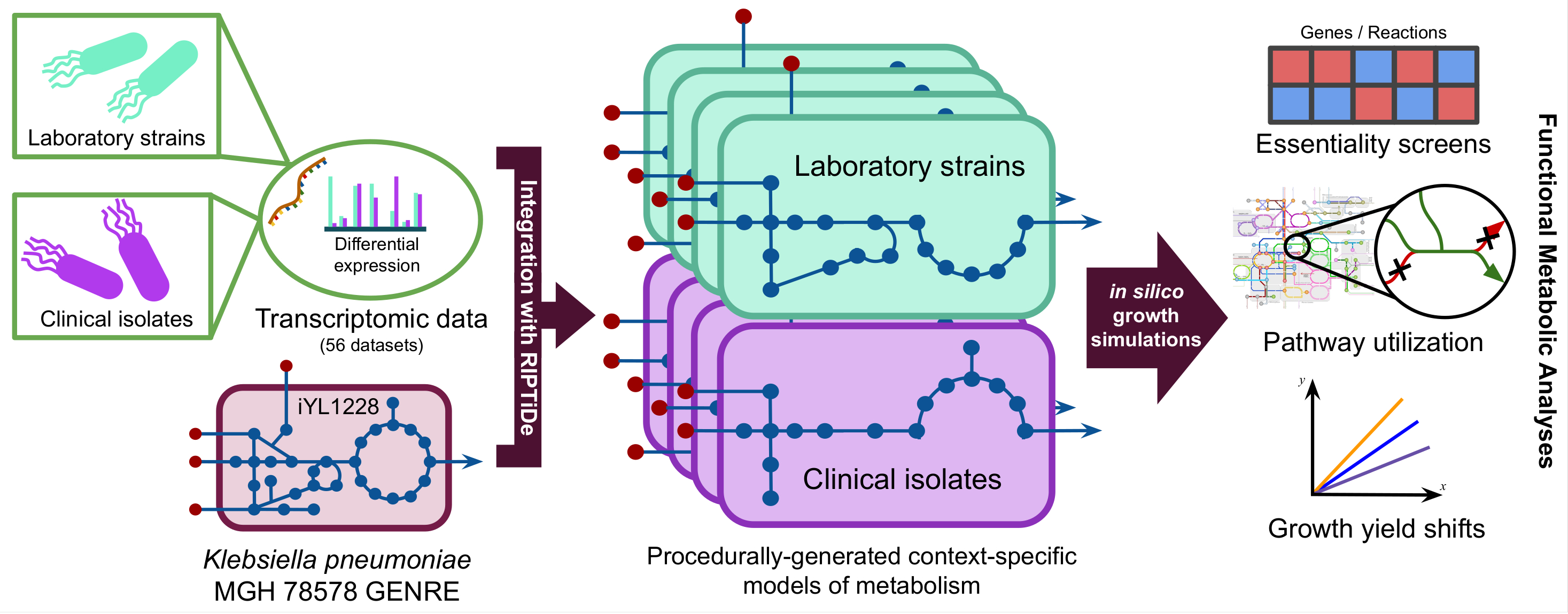
In our new pre-print, we ultimately found that clinical isolate-associated metabolic models were more dependent on environmental valine and consumed it at much faster rates that lab isolates in silico.